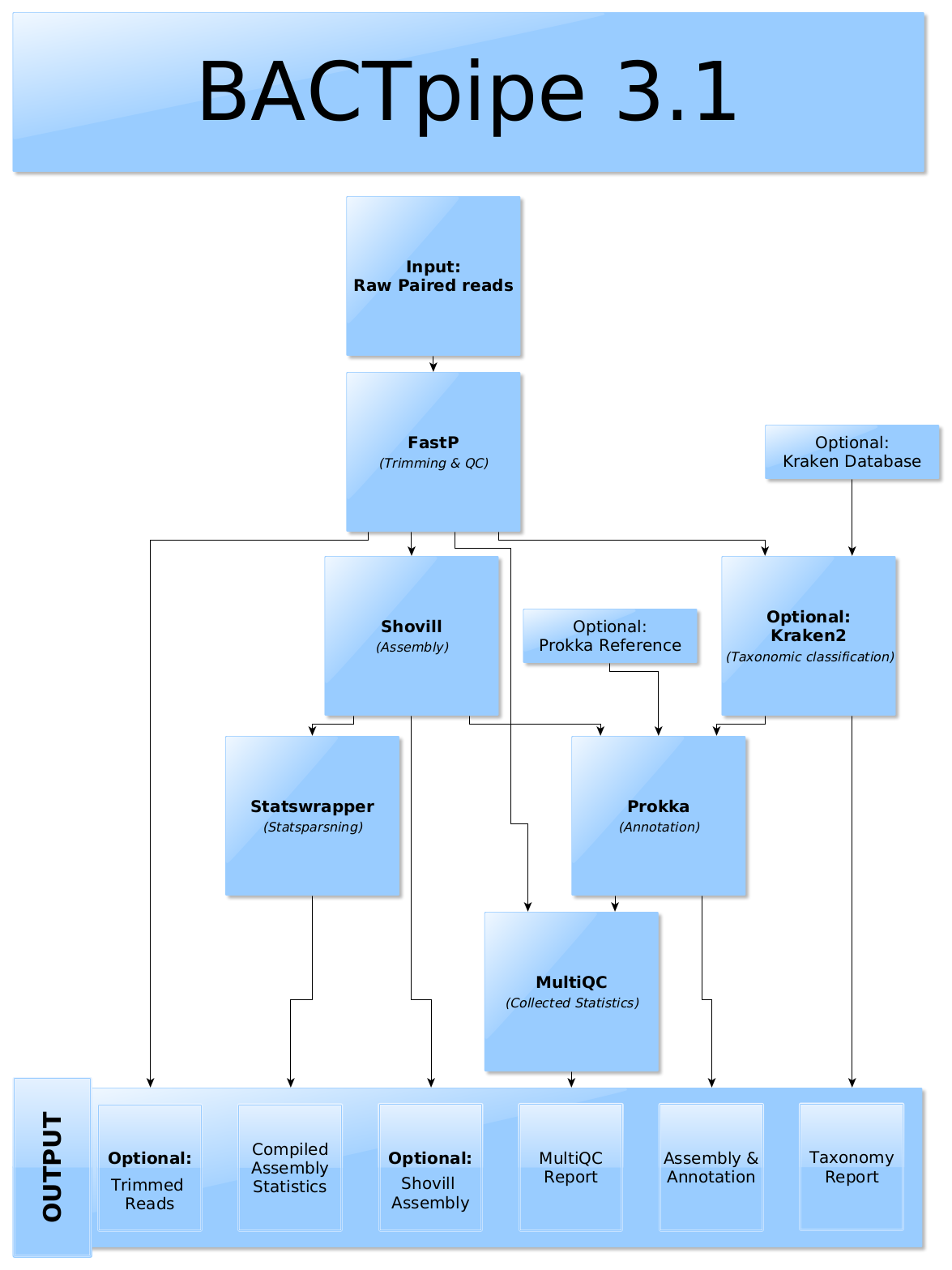
BACTpipe introduction and overview¶
BACTpipe uses whole genome shotgun sequenced, paired end reads, to assemble and annotate single bacterial genomes.
BACTpipe’s analysis flow starts with pre-processing of paired end reads in
fastq format using fastp, followed by taxonomic classification and gram
stain identification by Kraken2. This step also identifies if the sample is
potentially contaminated, i.e. contains more than one species. Then the sample
is de-novo assembled using Shovill. The draft genome fasta file headers
are renamed to get unique genome-specific headers.
Finally, genome annotation is performed using prokka with genus, species,
and gram stain information (if possible to uniquely identify in the Kraken2
step) added. Lastly, basic statistics about the assembly and annotation are
collected into a HTML report using MultiQC.
BACTpipe is implemented in Nextflow and an overview of the workflow can be seen below with the different output files at the bottom.